






Żeby to zobrazować powyżej pokazujemy obraz z kamery optycznej wbudowanego mikroskopu z celownikiem promieni rentgenowskich. Jeden z naszych klientów zajmujących się recyklingiem dostarczył nam próbkę układu scalonego by zbadać z jakich metali jest zbudowany. Dzięki precyzji tej maszyny możemy dokładnie określić skład pierwiastkowy nawet tak małych elementów. Na tym etapie nie będziemy się rozpisywać o możliwościach i próbkach bo to cały, oddzielny rozdział i o nim później. Skończymy teraz opis części sprzętowej i zajmiemy się teorią badania.

Rys. 11 Tankowanie 100 litrów ciekłego azotu do ciśnieniowego naczynia Dewara.
Zanim ją skończymy wrócimy jeszcze na chwilę do ciekłego azotu. W zestawie z maszyną było duże, ciśnieniowe naczynie Dewara o pojemności 100 litrów. Też go oczywiście kupiliśmy i to była dobra decyzja bo ciekły azot w takim ciśnieniowym zbiorniku dużo dłużej się utrzymuje niż w zwykłym, zamykanym styropianowym korkiem. W tym zbiorniku utrzymuje się stałe ciśnienie około 1,5 bara i co jakiś czas zawór zrzuca jego nadmiar powstały w wyniku parowania. Na powyższym zdjęciu pokazaliśmy proces tankowania azotu do tego zbiornika. Taki zbiornik ma jeszcze jedną zaletę a mianowicie łatwy sposób pobierania - dzięki nadciśnieniu wewnątrz oraz układowi wymuszania ciśnienia z parownikiem, możemy odkręcając jeden zawór, nalewać ciekły azot z węża.

Rys. 12 Uzupełnianie ciekłego azotu w naczyniu detektora.
Jak już wspomniano, zbiornik detektora zespolony z nim i umieszczony w maszynie ma pojemność 3 litrów, jest to taki sam, jak każdy inny zbiornik Dewara z izolacją próżniową i tak jak z każdego innego naczynia azot będzie parował. W przypadku naszej maszyny musimy wlewać do zbiornika około 1 litr azotu dziennie. Służy do tego specjalny lejek z rurką nadmiarową przez którą na zewnątrz wypłynie nadmiar azotu i nie zaleje całego urządzenia. Detektor posiada jeszcze czujnik temperatury który nie pozwoli uruchomić jego pracy, jeśli temperatura nie jest odpowiednia i stabilna. Musimy po prostu codziennie rano wlać do maszyny około litr cieczy by nie dopuścić do jego całkowitego wyparowania. Producent zaleca odczekanie 3 godzin po pierwszym schłodzeniu detektora dla zapewnienia stabilnych warunków pracy. Niestety, konieczność zużywania sporej ilości ciekłego azotu znacząco przyczynia się do kalkulacji końcowej każdego wykonywanego przez nas badania.
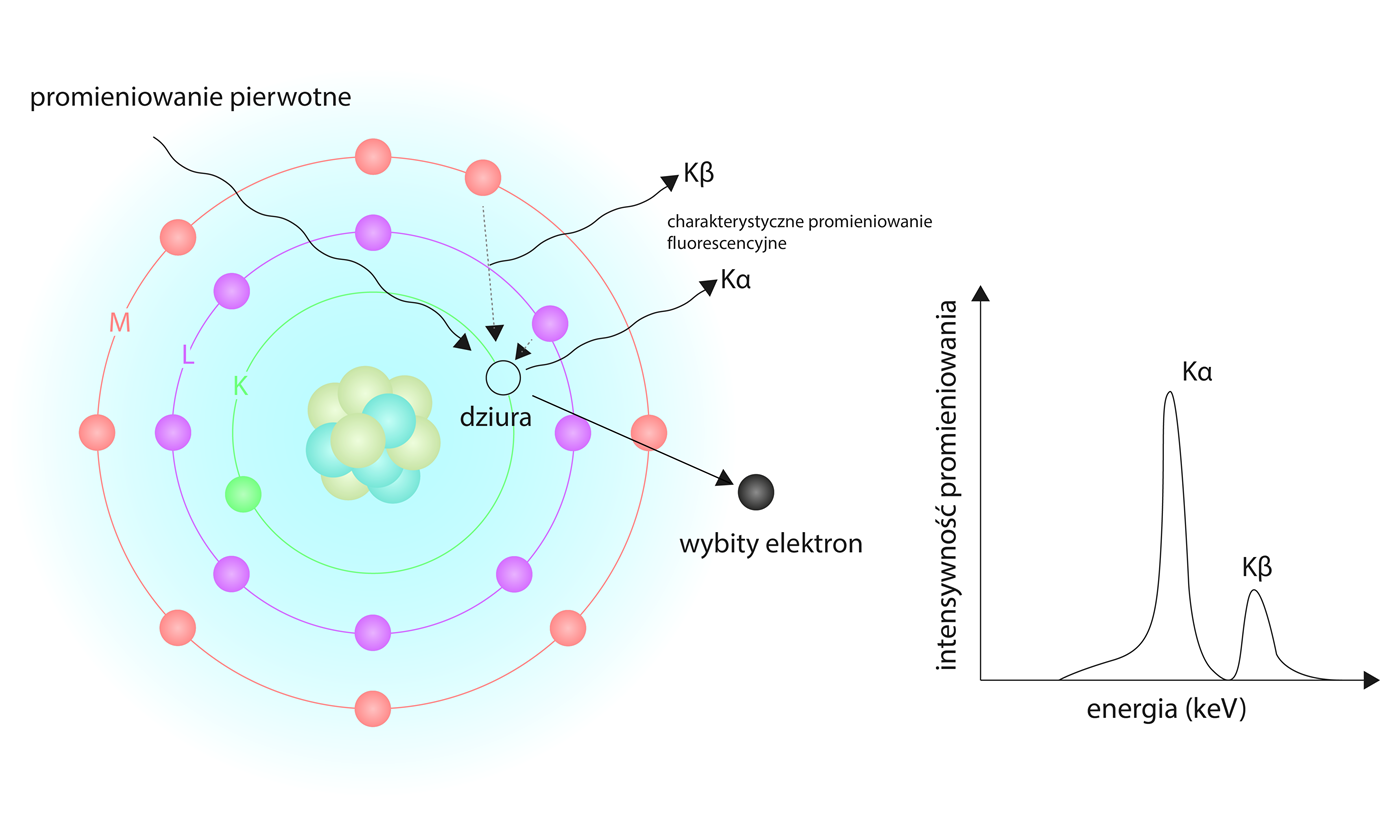
Rys. 13 Model ideowy badania XRF.
Na razie tyle jeśli chodzi o hardware. Zmieniamy skalę i przechodzimy na poziom atomu, jeśli kogoś idea badania nie interesuje może ten artykuł trochę przewinąć, dla wytrwałych prezentujemy powyższy model atomu. By zrozumieć ideę naszego badania musimy wiedzieć, że promieniowanie jonizujące wzięło swoją nazwę od swej natury czyli zdolności jonizowania atomów. Takim promieniowaniem jonizującym w naszym przypadku jest Promieniowanie Rentgena, ma ono na tyle dużą energię, by jego kwant wybijał elektrony atomów z ich naturalnych powłok. Wspomniane powłoki to orbity dla elektronów o kwantowej, różnej wartości energii. Od najniższej nazywamy je: K, L i M, jest ich więcej dla cięższych pierwiastków, jednak nas tamte raczej nie będą interesowały. Gdy kwant pierwotnego promieniowania z lampy, którą wcześniej się zachwycaliśmy w kontekście jej drogiej anody, wpadnie w atom - wybije elektrony z najniższej powłoki (K). Wynikający z tego brak elektronu w powłoce K zmusza jedną z powłok o wyższej energii (L lub M) do wypełnienia pustki, przywracając w ten sposób atom do stanu o najniższej energii. Gdy elektron opada na najgłębszą powłokę, musi uwolnić kwant energii równoważny różnicy energii między tymi dwiema powłokami - to będzie nasze promieniowanie fluorescencyjne.
Z pewnością prawdą jest, że atomy mają więcej niż jedną powłokę elektronową, z których każda zawiera elektrony, które są w stanie wypełnić pustą przestrzeń pozostawioną przez wybity elektron w najbardziej wewnętrznej powłoce. Wraz ze wzrostem liczby atomowej w układzie okresowym pierwiastków rośnie liczba elektronów, które pierwiastek może utrzymać na swoich orbitach w pewnej odległości. Tak więc, gdy elektron z powłoki K zostanie wybity, elektron z powłoki L lub M lub dowolnej kolejnej może zająć jego miejsce i dać sygnał fluorescencyjny. Elektrony, które spadają z powłoki L na powłokę K, dają energię oznaczoną jako Kα, podczas gdy te, które spadają z powłoki M, są określane jako Kβ. To właśnie tą energię wtórnego promieniowania rejestruje opisany wcześniej, chłodzony detektor. Procesor ją zbiera, kwantyfikuje, analizuje i rejestruje w postaci widma.
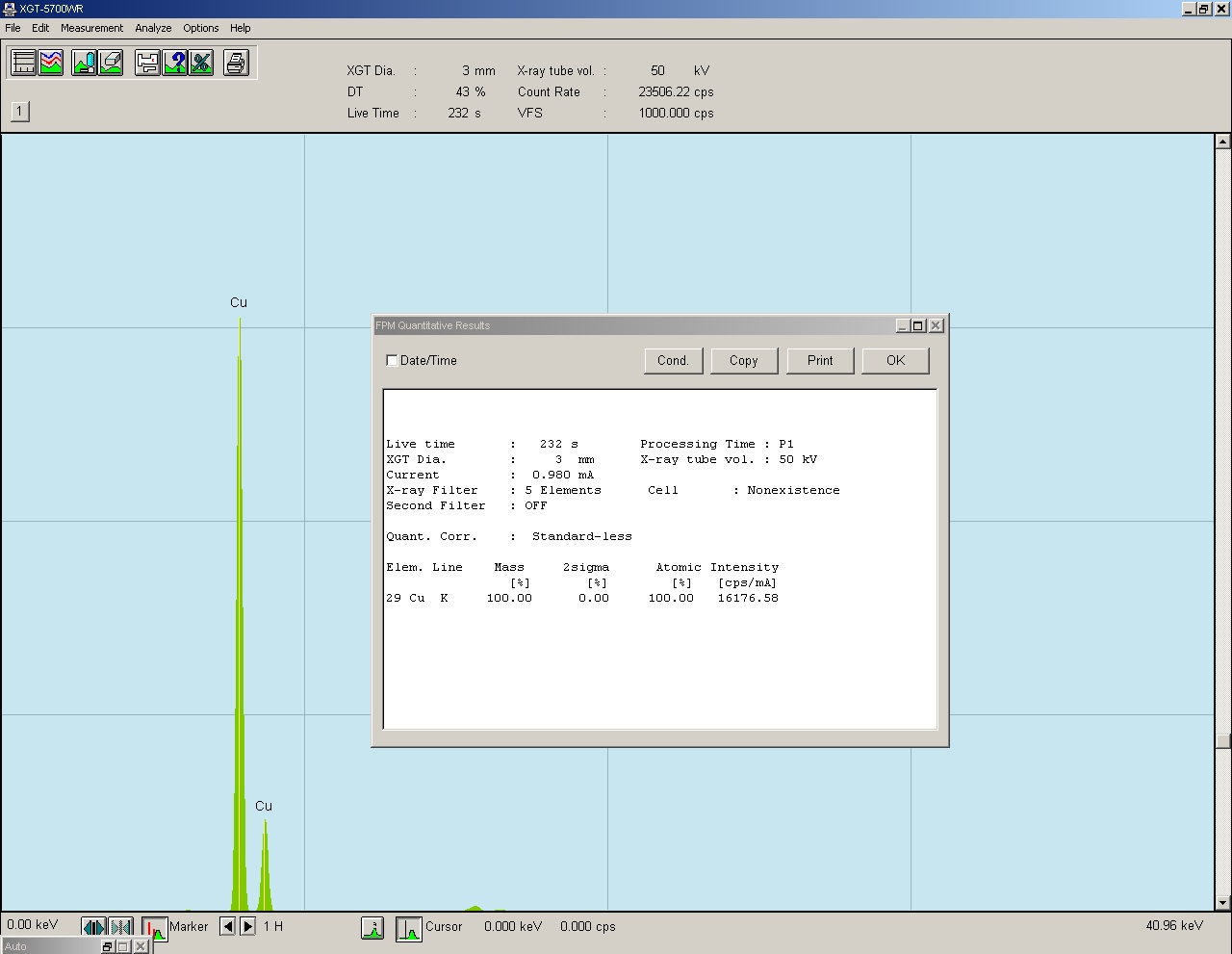
Rys. 14 Widmo energii zarejestrowanego promieniowania fluorescencyjnego.
Jak to wygląda w praktyce? - powyżej widmo z naszej maszyny dla analizy czystej miedzi, taki diagram dostaje operator po zbadaniu próbki. Na osi X mamy energię w keV a na osy Y intensywność zliczeń cps (zliczenia na sekundę). Warto w tym miejscu dodać, że istnieją dwa typy fluorescencyjnej analizy rentgenowskiej. Nasza maszyna reprezentuje metodę XRF-ED (fluorescencyjna spektrometria rentgenowska z dyspersją energii), drugą metodą jest XRF-WD ( fluorescencyjna spektrometria rentgenowska z dyspersją długości fali), ta metoda jest dużo bardziej skomplikowana i opiera się na kątach padania fal i ich interferencji.
Wracając do naszej metody i widma, zobaczmy co dzieje się podczas prowadzenia analizy - wprowadzamy naszą próbkę do maszyny i zaczynamy ją oświetlać promieniami rentgena. Próbka cały czas oddaje wtórne promieniowanie, które pada na detektor, sygnały z detektora są mierzone i dorzucane na diagram w miejsca, gdzie reprezentują daną energię. Na początku pojawia się trochę szumu na całej długości osi X i zaczynają wyłaniać się poszczególne piki. Rosną w trakcie całego pomiaru, trwającego w zależności od nastaw, maksymalnie 2 minuty lub dłużej jeśli mamy do czynienia z lekkimi pierwiastkami i niskimi ich zawartościami. Po zakończeniu pomiaru do pracy bierze się już tylko oprogramowanie komputera, które w oparciu o wcześniej skalibrowane matryce rozpoznaje jakie pierwiastki dały konkretny sygnał i oblicza ich zawartości. I tu znów można otworzyć kolejne drzwi do nowego rozdziału poświęconego obliczeniom stężeń. Nasza maszyna oferuje dwie metody oznaczania stężeń: metodę podstawowych parametrów FPM oraz analizę w oparciu o krzywe kalibracyjne.
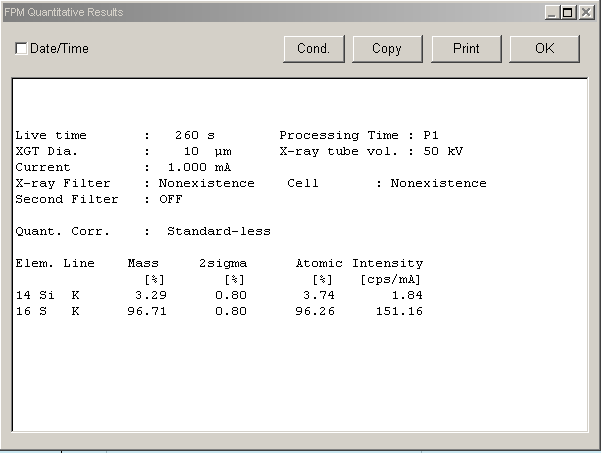
Rys. 15 Wynik ilościowy analizy obliczony metodą FPM.
Metoda FPM jest najprostsza i w wielu badaniach świetnie się sprawdza, nowoczesne oprogramowanie samo rozpoznaje położenia pików, ich intensywności i charakterystyczne wzorce z różnych linii emisji. Tak, jak powyżej, gdzie mamy wynik z badania czystej siarki. Urządzenie oprócz tego pierwiastka wykryło w nim 3,29% krzemu. W widmie mogą też być obecne pewne piki artefaktów, w tym charakterystyczne linie rozproszone Rayleigha i Comptona z generatora promieniowania rentgenowskiego, piki spowodowane dyfrakcją promieniowania rentgenowskiego oraz piki sumy/ucieczki. Ich znajomość jest konieczna, aby uniknąć błędnej interpretacji wyników. Dlatego uznaliśmy, że w tym artykule nie będziemy już głębiej wnikać w szczegóły a zrobimy to w kolejnych artykułach poświęconych konkretnym analizom, nie poruszymy tu już też drugiej metody obliczeń w oparciu o krzywe kalibracyjne, która zapewnia niesamowicie małe wykrywalności i oznaczania substancji od ppm do 100%.

Rys. 16 Przygotowanie próbki siarki do analizy.
Na przykładzie badania czystej siarki, którego wynik mogliśmy zobaczyć, przejdziemy do innego, acz bardzo ważnego w tych badaniach aspektu, mowa tu o preparatyce próbek. O ile z litymi metalami, ceramiką czy tworzywami sztucznymi nie ma problemu bo wystarczy niewielki kawałek takiego elementu położyć na stolik pomiarowy, o tyle w przypadku proszków lub materiałów niejednolitych musimy się bardziej postarać. Gdy chcieliśmy zbadać siarkę z naszego słoja z odczynnikiem nie mogliśmy od tak, wysypać jej odrobiny na stolik pomiarowy. Ciężko było by ustawić odpowiednią odległość głowicy pomiarowej no i wewnątrz maszyny jest spora ilość wentylatorów chłodzących poszczególne elementy, które po prostu zdmuchnęły by próbkę zanieczyszczając jednocześnie całą maszynę. W przypadku takich proszków można zastosować dwie metody preparatyki próbki: stapianie lub prasowanie.

Rys. 17 Przygotowanie próbki sproszkowanego złota.
W przypadku siarki wystarczyło po prostu stopić próbkę co zapewniło jej zwartą i jednorodną strukturę, którą już w łatwy sposób można oświetlić wiązką. Jeśli stopienie jest nie możliwe, lub mogło by spowodować uszkodzenie materiału stosuje się prasowanie próbki do postaci tabletki. W tym przypadku klient dostarczył nam próbkę złota po chemicznym strąceniu z roztworu. Jego rozdrobnienie było niezwykle duże, najmniejszy podmuch powietrza powodował roznoszenie się pyłu.

Rys. 18 Proszek złota po chemicznym strąceniu z roztworu.
W takim przypadku możemy zastosować pastylkarkę oraz widoczną na poniższym zdjęciu hydrauliczną prasę zgniatającą o nacisku aż 100 ton. Nasza pastylkarka to urządzenie specjalnie wykonane do prasowania pastylek analitycznych pod wysokim ciśnieniem. Pomiędzy dwa walce wykonane z bardzo twardego spieku wsypuje się proszek i zgniata w stalowej tulei. Bardzo duże ciśnienie oddziałujące na materiał powoduje, że proszki zamieniają się w praktycznie jednolity, trwały materiał - idealny do badania naszą metodą. Jak widać na zdjęciu Nr 17 pastylka wykonana z proszku złota praktycznie niczym nie różni się od tłoczonej z metalu złotej monety.

Rys. 19 Hydrauliczna prasa o nacisku 100 ton.
Pracująca w naszym laboratorium prasa stosowana była do badań wytrzymałościowych betonu, jednak świetnie sprawdza się jako urządzenie do ściskania naszej pastylkarki. Jest to połączenie małego tłoczka, którym zwiększamy ciśnienie w tłoku zgniatającym o dużej średnicy, małym tłoczkiem trzeba się trochę namachać jednak dzięki temu uzyskiwane siły są bardzo duże. Jest to ważne gdy np. chcemy sprasować materiał nie mający chęci do połączenia się w jednolitą strukturę jak np.jedna z próbek osadu z komór do napylania metali naszego klienta, która w większości miała składać się ze srebra. W tym miejscu warto wspomnieć o metodzie uśredniania i przygotowania proszków do prasowania.

Rys. 20 Próbka zmielona w młynie planetarno - kulowym, przesiana i sprasowana.










