







Analiza składu chemicznego materiałów jest kluczowym elementem w dziedzinie nauki, technologii i produkcji. Zazwyczaj wymaga skomplikowanych i inwazyjnych metod chemicznych, dających konkretne efekty w reakcjach badanych substancji z innymi. Wymaga to dużej ilości bardzo czystych odczynników, bardzo dużej ilości pracy i reakcji by znaleźć oraz określić stężenia konkretnych pierwiastków. W tym kontekście tytułowa spektrometria fluorescencji rentgenowskiej (XRF) jest niezwykle przydatną techniką. Wykorzystuje ona promieniowanie rentgenowskie do pobudzania atomów próbek. To pozwala na identyfikację i ilościowe określenie składu pierwiastkowego na podstawie rejestracji i analizy wtórnego promieniowania, które jest wysyłane przez atomy gdy powracają do swojego podstawowego stanu. Dzięki temu możemy badać różnorodne próbki, takie jak metale, minerały, materiały ceramiczne czy nawet substancje rozpuszczone w roztworach, w sposób całkowicie bezinwazyjny, bez zużycia ogromnej ilości odczynników i powstawania jakichkolwiek odpadów.
Niniejszy artykuł zaprezentuje zasadę działania tej maszyny, podstawy i istotę samej analizy oraz przede wszystkim nasze zmagania z uruchomieniem i kalibracją.
Nasza maszyna to produkt renomowanej w branży firmy HORIBA z Francji, jej symbol: XGT-5700WR. Jest to spektrometr do analizy jakościowej i ilościowej pierwiastków ze szczególnym naciskiem na dyrektywę WEEE/RoHS, która to dotyczy zawartości metali ciężkich w sprzęcie elektronicznym (Pb/Cd/Cr/Hg/Br). Zastosowano kilka innowacji, aby osiągnąć bardzo dobrą czułość dla wspomnianych wcześniej 5 pierwiastków w szerokim zakresie zawartości, jednak to nie sprawia, że maszyna jest dedykowana tylko pod tą analizę. Świetnie radzi sobie z analizą pierwiastków zarówno lekkich jak i ciężkich. Oczywiście nie wszystkich, bo jej czułość w przypadku pierwiastków lekkich zaczyna się dopiero od sodu. To sprawia wiele problemów związanych z ilościowym określaniem zawartości. Bo jeśli badamy np. próbkę materii organicznej to jak uwzględnić niewidoczny węgiel, azot czy tlen? O tym wszystkim napiszemy w dalszej części artykułu, jednak widać już, że taka maszyna to nie wszechmocne panaceum na wszystkie znoje chemii analitycznej :). 
Rys. 1 Stanowisko pracy ze spektrometrem XRF.
Panaceum na pewno nie, ale i tak ma potężne możliwości w porównaniu do tradycyjnych metod analitycznych. Na powyższym zdjęciu pokazaliśmy całe stanowisko pracy maszyny w naszym laboratorium. Największy element to główna maszyna pomiarowa. To tutaj znajduje się źródło promieni rentgenowskich, detektor promieniowania wtórnego, komora badawcza do której wprowadza się próbkę, kamery, oświetlacze, mikroskop optyczny i wiele innych. Za chwilę rozwiniemy dwie pozycje z tych elementów czyli detektor promieni wtórnych i komorę pomiarową. Obok głównej maszyny pomiarowej znajduje się jednostka procesora (jak to nazywa producent) - tam mieści się wysoko precyzyjny zasilacz wysokiego napięcia do zasilania lampy rentgenowskiej oraz układ akwizycji i przetwarzania danych napływających z detektora. Obie te części połączone są całkiem sporą ilością przewodów. Wszystko sterowane jest z komputera PC za pomocą dedykowanej karty PCI.

Rys. 2 Podłączanie pompy próżniowej do systemu.
Powyższe zdjęcie przedstawia podłączanie kolejnego ważnego elementu systemu - jest to pompa próżniowa, która służy do opróżniania toru promieni rentgenowskich z powietrza dla poprawy jakości pomiaru. Część promieni rentgenowskich na pewno oddziaływała by z atomami powietrza osłabiając ich intensywność i na pewno też wprowadzając błędne sygnały na detektor. W tym zadaniu pomagał mój syn Wiktor.

Rys. 3 Połączenia elektryczne obu części maszyny analitycznej XRF.
Wracając do wspomnianego przed chwilą rozwinięcia, w pierwszym przypadku rozwinięcie to związane jest z białą bańką którą widać pod stołem - tak to naczynie Dewara na ciekły azot. Jego związek z detektorem jest taki, że dla zapewnienia dobrego stosunku sygnału do szumu detektor schłodzony musi być do temperatury -196 stopni Celsjusza. W maszynie zastosowano bardzo wysokiej jakości detektor krzemowy.
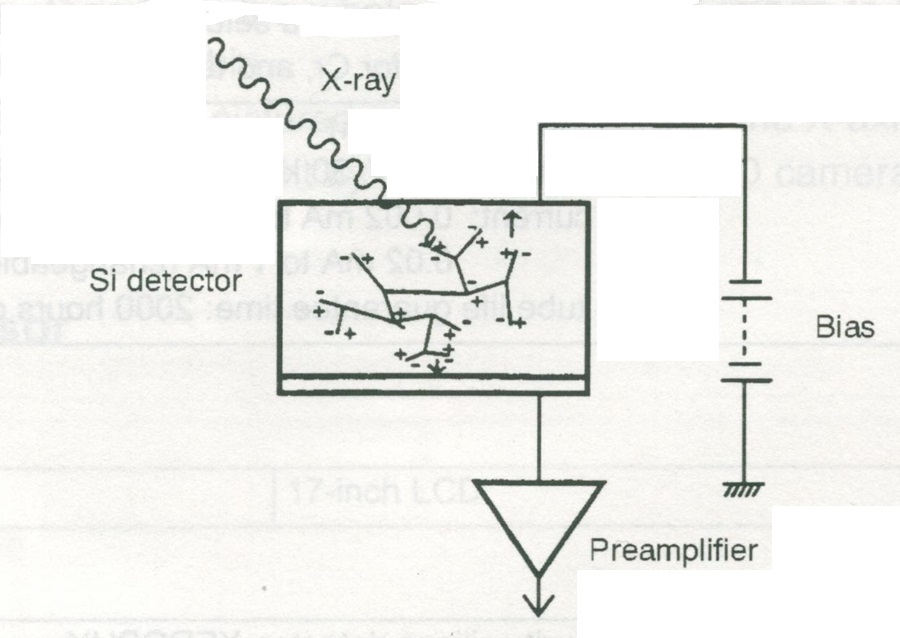
Rys. 4 Schemat detektora krzemowego.
Schłodzony ciekłym azotem półprzewodnik krzemowy spolaryzowany jest w kierunku zaporowym przez zewnętrzne napięcie. Gdy w obszar detektora wpadnie kwant promieniowania wtórnego z próbki, powstaje krótkotrwały impuls prądu proporcjonalny do energii promieniowania. Dalej ten impuls trafia na przedwzmacniacz i zamieniany jest na napięcie elektryczne. Wtedy pracę przejmuje wielokanałowy analizator amplitud, który klasyfikuje i liczy wszystkie powstałe impulsy tworząc widmo ilości zliczeń pików o danym napięciu. Co z tym dalej się dzieje pokażemy w dalszej części opracowania.

Rys. 5 Widok detektora krzemowego z osprzętem.
Naszej maszyny nie kupiliśmy, jak to się mówi prosto z salonu, bo nie było by nas na coś takiego po prostu stać. Kupiliśmy ją na aukcji przemysłowej z fabryki produkującej elektronikę. W zestawie dostaliśmy także wadliwy detektor po wymianie serwisowej, dzięki czemu możemy pokazać jego budowę i wszystkie opisane wcześniej elementy. Biała bańka to naczynie Dewara o pojemności 3 litrów - jej wlot mieści się na szczycie maszyny analitycznej by w prosty sposób można ją było uzupełniać. Kolejne widoczne elementy to zasilacz polaryzujący detektor, przedwzmacniacz sygnału z detektora oraz głowica pomiarowa, która znajduje się blisko próbki by "zbierać" wtórne promieniowanie.
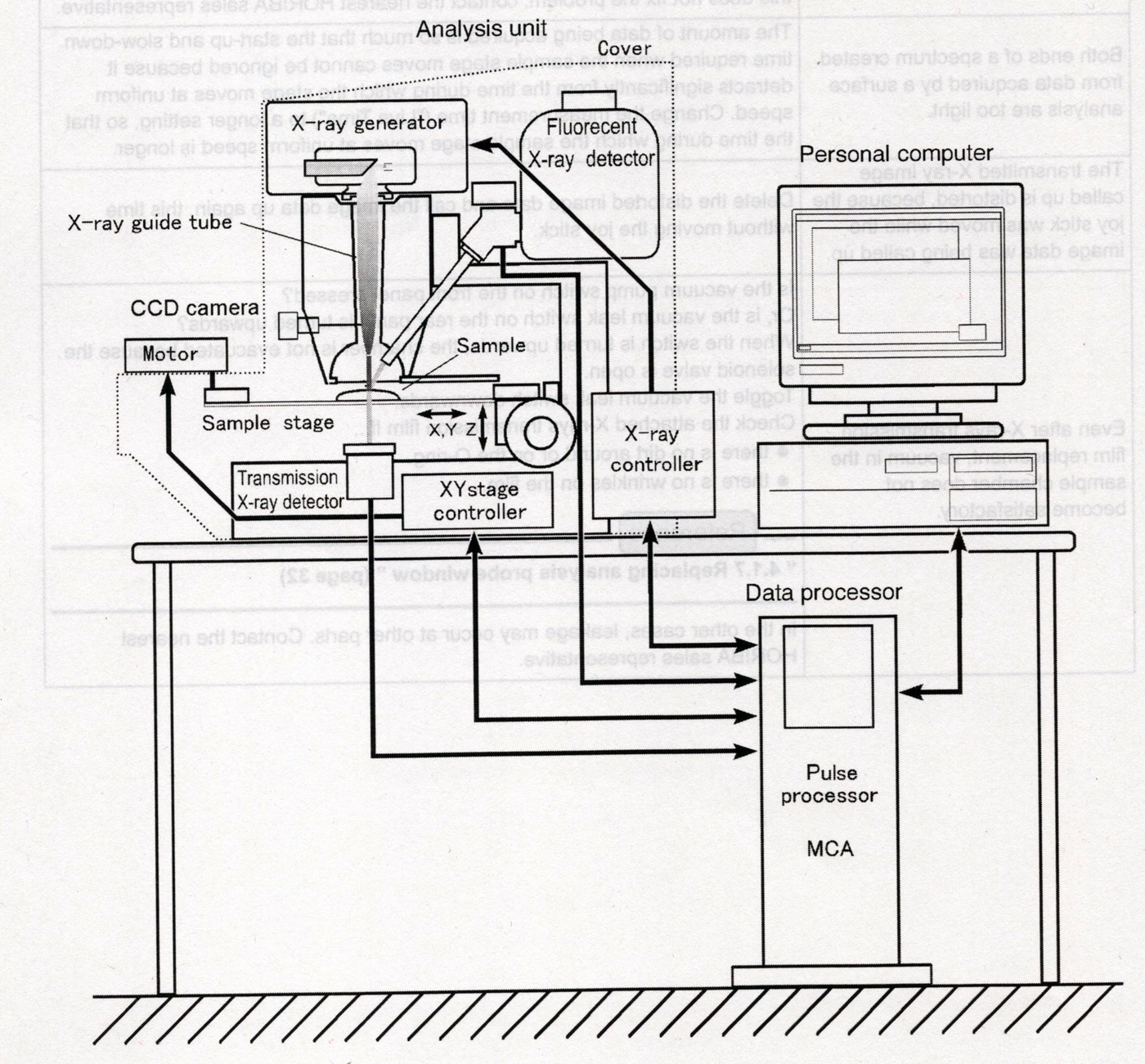
Rys. 6 Diagram blokowy całego systemu XRF.
Teraz prezentujemy kolejny skan z dokumentacji technicznej maszyny. Tutaj dobrze pokazano umiejscawianie detektora o którym jest mowa. Jego głowica znajduje się zaraz nad próbką by wychwytywać wtórne promieniowanie rentgenowskie, jest ono bardzo ważne, dlatego w dalszej części artykułu jeszcze sporo o nim napiszemy. Napisaliśmy, że rozwiniemy opis jeszcze jednej części głównej maszyny pomiarowej a mianowicie komory dla próbek. Powyższa ilustracja już pokazuje dlaczego nieco więcej chcemy o tym napisać. Jest tam stoliczek na próbki, jednak nie taki zwykły - posiada precyzyjny, pozycjonowany napęd w osiach X i Y sterowany numerycznie z komputera. Daje to dwie znakomite możliwości maszynie. Po pierwsze, w zestawie z kamerą i mikroskopem optycznym, możemy bardzo precyzyjnie ustawić miejsce w które wycelujemy promienie rentgena i zbadamy je a po drugie, z funkcją systemu możemy skanować duży obszar próbki i wykonać mapowanie pierwiastkowe.
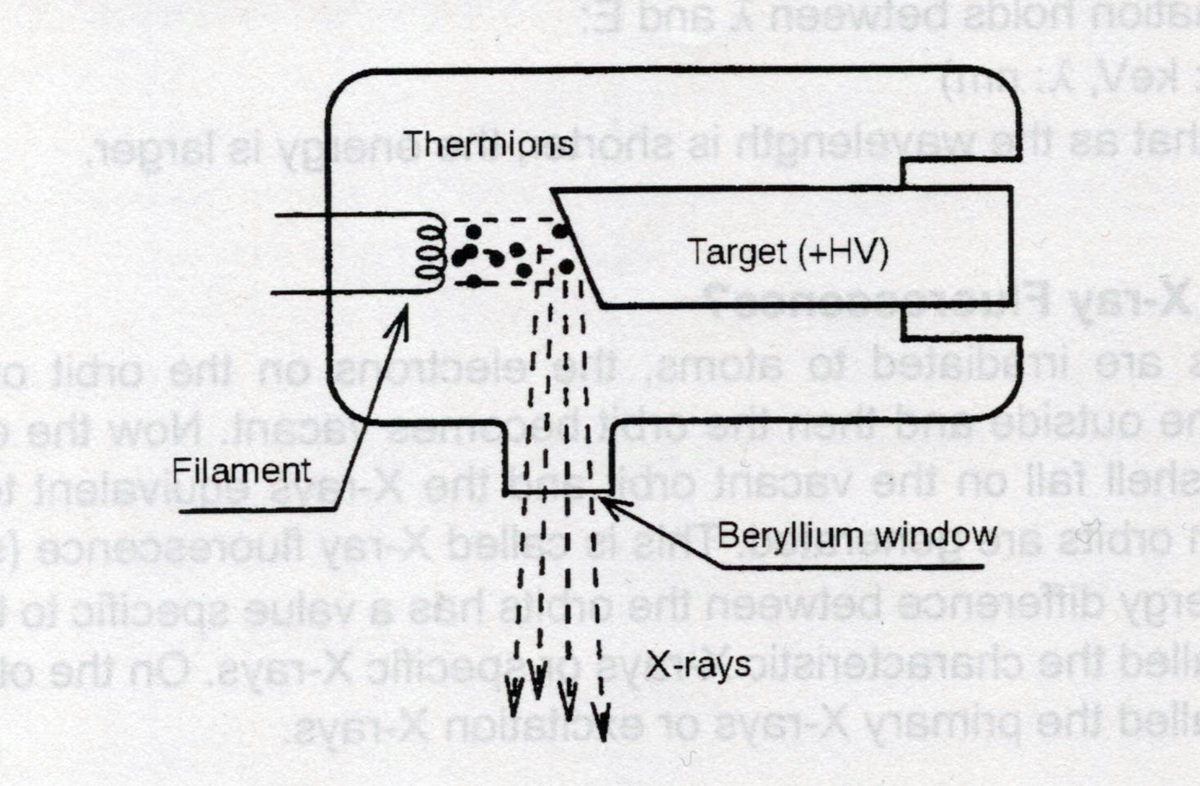
Rys. 7 Diagram blokowy źródła promieni Rentgena.
W tej części artykułu dotyczącej sprzętu musimy opisać jeszcze jeden, bardzo ważny element - źródło promieni X - lampę rentgenowską. Jest to urządzenie zasadą działania identyczne do każdej innej lampy: rozgrzany przez płynący prąd żarzenia wolframowy filament wysyła w próżni elektrony, które są przyśpieszane różnicą potencjału do anody. Uderzając w nią gwałtownie hamują co jest źródłem promieniowania. Różnic w stosunku do np. medycznych lamp jest wiele, jednak chyba najważniejszą jest materiał anody, bo od niego zależy jakość i energia promieniowania. W tej lampie zastosowano czysty rod - bardzo drogi pierwiastek ziem rzadkich.

Rys. 8 Widok źródła promieni Rentgena zamontowany w maszynie.
Materiałem obudowy nie jest szkło a ceramika z dodatkową obudową metalową a promienie wychodzą przez bardzo malutką głowicę z okienkiem berylowym wprost w próżniowy tunel z przesłonami i filtrami. Obudowa dodatkowo wypełniona jest olejem i warstwą ołowiu dla izolacji promieniowania w innych kierunkach dla ochrony obsługi. Stąd też duży wymiar elementu no i jego waga sięgająca prawie 15 kg. Lampa ta zasilana jest napięciem 15, 30 lub 50 kV a prąd lampy ustawiany jest bardzo precyzyjnie od 0,02 do 1 mA. Wszystkie zmiany nastaw prądu i napięcia lampy wykonywane są poprzez program na komputerze PC.

Rys. 9 Widok źródła promieni Rentgena poza maszyną - część zamienna.
Jest to tzw. źródło mikro-wiązkowe, dodatkowo w tunelu próżniowym znajduje się układ formowania wiązki dla zapewnienia precyzji pomiaru. Mikroskop oferuje dwie średnice wiązki: 3mm oraz 10µm. Ta większa stosowana jest do pomiaru lekkich pierwiastków - jest mniej precyzyjna jeśli chodzi o obszar pomiarowy ale za to daje lepsze intensywności emisji dla lekkich pierwiastków. Wiązka o średnicy 10µm pozwala bardzo precyzyjnie zbadać zawartość pierwiastków w bardzo małych elementach widocznych na ekranie już pod mikroskopem optycznym jak np. skład pierwiastkowy drucika łączącego pin ze strukturą układu scalonego.

Rys. 10 Obraz z mikroskopu optycznego - ustalanie punktu pomiaru.
Żeby to zobrazować powyżej pokazujemy obraz z kamery optycznej wbudowanego mikroskopu z celownikiem promieni rentgenowskich. Jeden z naszych klientów zajmujących się recyklingiem dostarczył nam próbkę układu scalonego by zbadać z jakich metali jest zbudowany. Dzięki precyzji tej maszyny możemy dokładnie określić skład pierwiastkowy nawet tak małych elementów. Na tym etapie nie będziemy się rozpisywać o możliwościach i próbkach bo to cały, oddzielny rozdział i o nim później. Skończymy teraz opis części sprzętowej i zajmiemy się teorią badania.

Rys. 11 Tankowanie 100 litrów ciekłego azotu do ciśnieniowego naczynia Dewara.
Zanim ją skończymy wrócimy jeszcze na chwilę do ciekłego azotu. W zestawie z maszyną było duże, ciśnieniowe naczynie Dewara o pojemności 100 litrów. Też go oczywiście kupiliśmy i to była dobra decyzja bo ciekły azot w takim ciśnieniowym zbiorniku dużo dłużej się utrzymuje niż w zwykłym, zamykanym styropianowym korkiem. W tym zbiorniku utrzymuje się stałe ciśnienie około 1,5 bara i co jakiś czas zawór zrzuca jego nadmiar powstały w wyniku parowania. Na powyższym zdjęciu pokazaliśmy proces tankowania azotu do tego zbiornika. Taki zbiornik ma jeszcze jedną zaletę a mianowicie łatwy sposób pobierania - dzięki nadciśnieniu wewnątrz oraz układowi wymuszania ciśnienia z parownikiem, możemy odkręcając jeden zawór, nalewać ciekły azot z węża.

Rys. 12 Uzupełnianie ciekłego azotu w naczyniu detektora.
Jak już wspomniano, zbiornik detektora zespolony z nim i umieszczony w maszynie ma pojemność 3 litrów, jest to taki sam, jak każdy inny zbiornik Dewara z izolacją próżniową i tak jak z każdego innego naczynia azot będzie parował. W przypadku naszej maszyny musimy wlewać do zbiornika około 1 litr azotu dziennie. Służy do tego specjalny lejek z rurką nadmiarową przez którą na zewnątrz wypłynie nadmiar azotu i nie zaleje całego urządzenia. Detektor posiada jeszcze czujnik temperatury który nie pozwoli uruchomić jego pracy, jeśli temperatura nie jest odpowiednia i stabilna. Musimy po prostu codziennie rano wlać do maszyny około litr cieczy by nie dopuścić do jego całkowitego wyparowania. Producent zaleca odczekanie 3 godzin po pierwszym schłodzeniu detektora dla zapewnienia stabilnych warunków pracy. Niestety, konieczność zużywania sporej ilości ciekłego azotu znacząco przyczynia się do kalkulacji końcowej każdego wykonywanego przez nas badania.
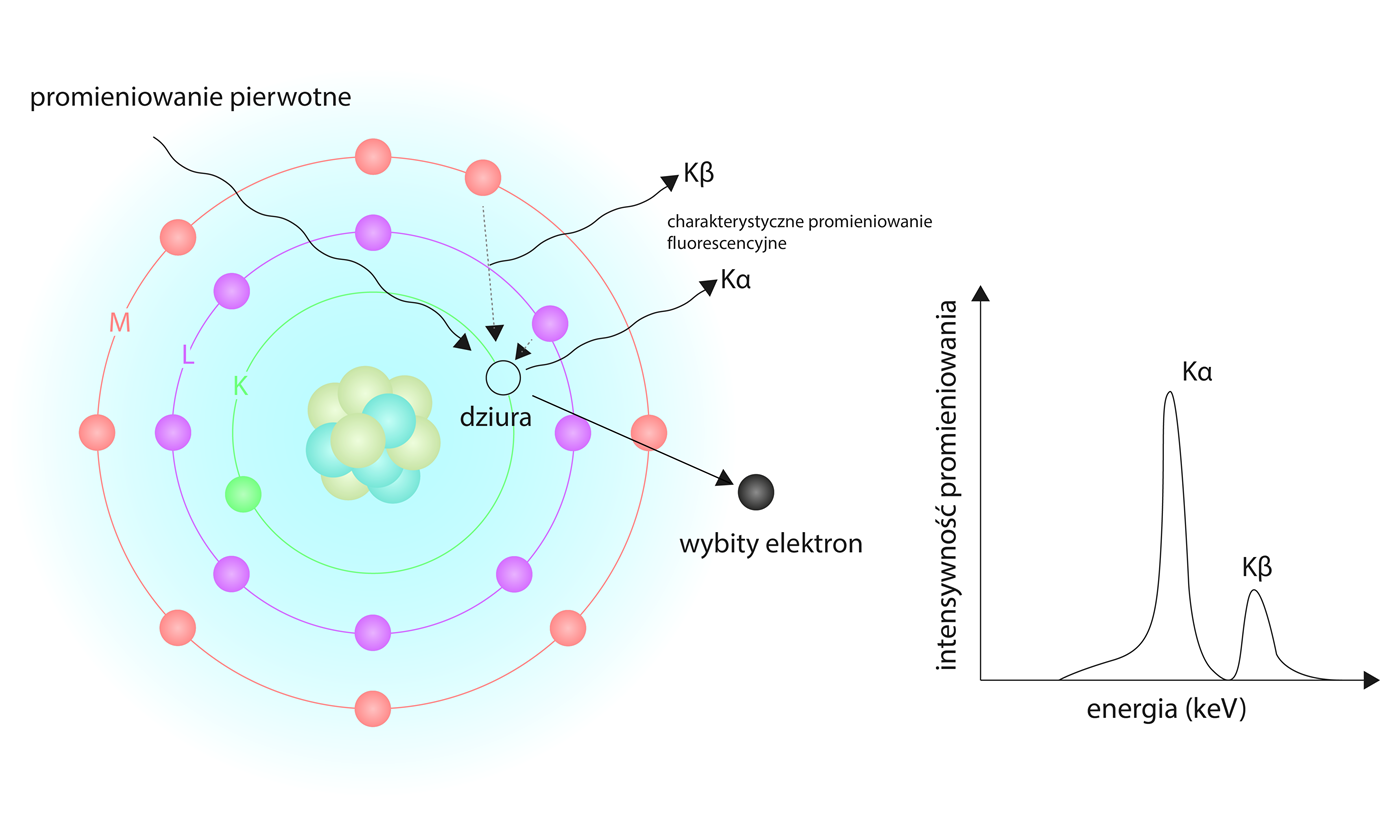
Rys. 13 Model ideowy badania XRF.
Na razie tyle jeśli chodzi o hardware. Zmieniamy skalę i przechodzimy na poziom atomu, jeśli kogoś idea badania nie interesuje może ten artykuł trochę przewinąć, dla wytrwałych prezentujemy powyższy model atomu. By zrozumieć ideę naszego badania musimy wiedzieć, że promieniowanie jonizujące wzięło swoją nazwę od swej natury czyli zdolności jonizowania atomów. Takim promieniowaniem jonizującym w naszym przypadku jest Promieniowanie Rentgena, ma ono na tyle dużą energię, by jego kwant wybijał elektrony atomów z ich naturalnych powłok. Wspomniane powłoki to orbity dla elektronów o kwantowej, różnej wartości energii. Od najniższej nazywamy je: K, L i M, jest ich więcej dla cięższych pierwiastków, jednak nas tamte raczej nie będą interesowały. Gdy kwant pierwotnego promieniowania z lampy, którą wcześniej się zachwycaliśmy w kontekście jej drogiej anody, wpadnie w atom - wybije elektrony z najniższej powłoki (K). Wynikający z tego brak elektronu w powłoce K zmusza jedną z powłok o wyższej energii (L lub M) do wypełnienia pustki, przywracając w ten sposób atom do stanu o najniższej energii. Gdy elektron opada na najgłębszą powłokę, musi uwolnić kwant energii równoważny różnicy energii między tymi dwiema powłokami - to będzie nasze promieniowanie fluorescencyjne.
Z pewnością prawdą jest, że atomy mają więcej niż jedną powłokę elektronową, z których każda zawiera elektrony, które są w stanie wypełnić pustą przestrzeń pozostawioną przez wybity elektron w najbardziej wewnętrznej powłoce. Wraz ze wzrostem liczby atomowej w układzie okresowym pierwiastków rośnie liczba elektronów, które pierwiastek może utrzymać na swoich orbitach w pewnej odległości. Tak więc, gdy elektron z powłoki K zostanie wybity, elektron z powłoki L lub M lub dowolnej kolejnej może zająć jego miejsce i dać sygnał fluorescencyjny. Elektrony, które spadają z powłoki L na powłokę K, dają energię oznaczoną jako Kα, podczas gdy te, które spadają z powłoki M, są określane jako Kβ. To właśnie tą energię wtórnego promieniowania rejestruje opisany wcześniej, chłodzony detektor. Procesor ją zbiera, kwantyfikuje, analizuje i rejestruje w postaci widma.
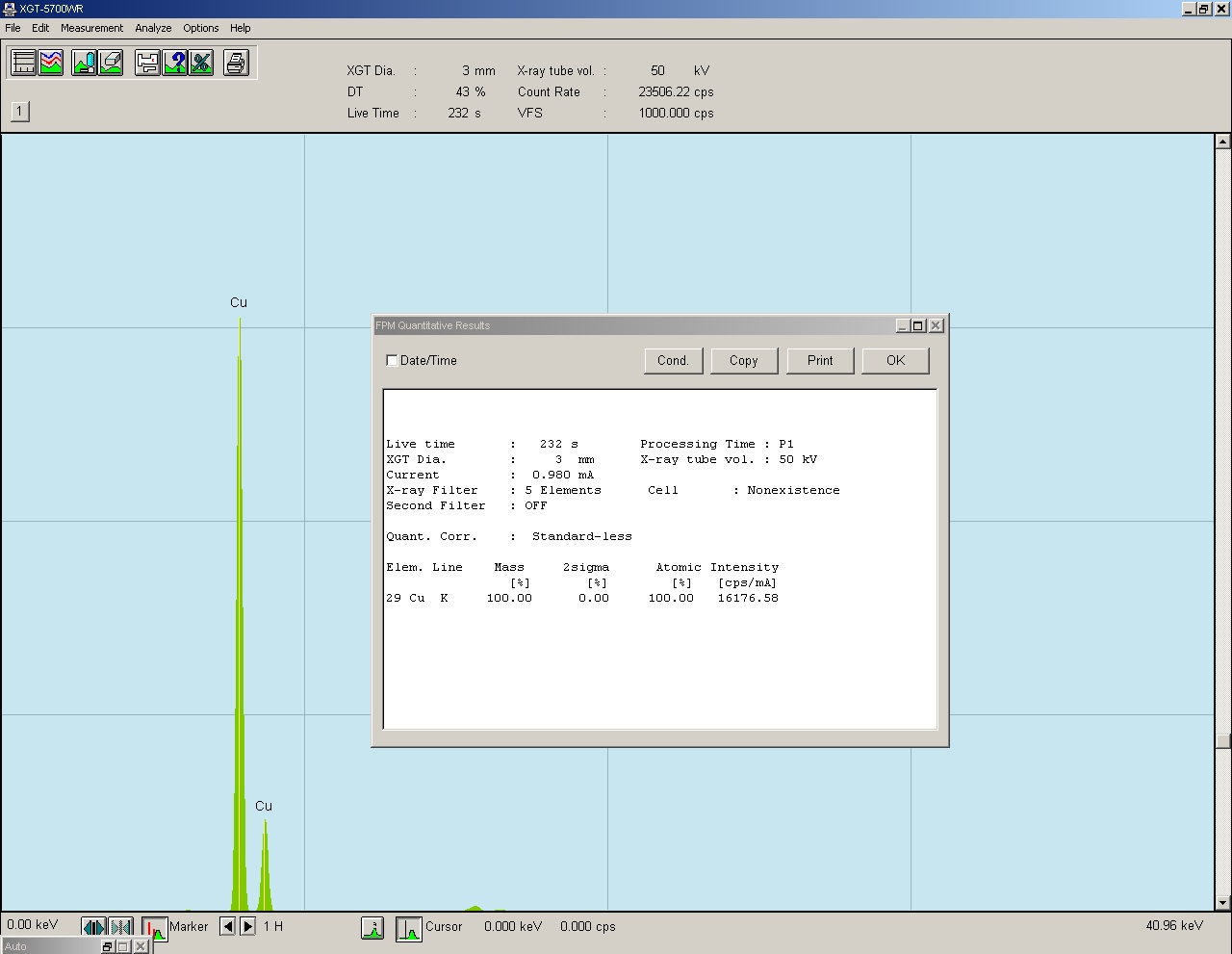
Rys. 14 Widmo energii zarejestrowanego promieniowania fluorescencyjnego.
Jak to wygląda w praktyce? - powyżej widmo z naszej maszyny dla analizy czystej miedzi, taki diagram dostaje operator po zbadaniu próbki. Na osi X mamy energię w keV a na osy Y intensywność zliczeń cps (zliczenia na sekundę). Warto w tym miejscu dodać, że istnieją dwa typy fluorescencyjnej analizy rentgenowskiej. Nasza maszyna reprezentuje metodę XRF-ED (fluorescencyjna spektrometria rentgenowska z dyspersją energii), drugą metodą jest XRF-WD ( fluorescencyjna spektrometria rentgenowska z dyspersją długości fali), ta metoda jest dużo bardziej skomplikowana i opiera się na kątach padania fal i ich interferencji.
Wracając do naszej metody i widma, zobaczmy co dzieje się podczas prowadzenia analizy - wprowadzamy naszą próbkę do maszyny i zaczynamy ją oświetlać promieniami rentgena. Próbka cały czas oddaje wtórne promieniowanie, które pada na detektor, sygnały z detektora są mierzone i dorzucane na diagram w miejsca, gdzie reprezentują daną energię. Na początku pojawia się trochę szumu na całej długości osi X i zaczynają wyłaniać się poszczególne piki. Rosną w trakcie całego pomiaru, trwającego w zależności od nastaw, maksymalnie 2 minuty lub dłużej jeśli mamy do czynienia z lekkimi pierwiastkami i niskimi ich zawartościami. Po zakończeniu pomiaru do pracy bierze się już tylko oprogramowanie komputera, które w oparciu o wcześniej skalibrowane matryce rozpoznaje jakie pierwiastki dały konkretny sygnał i oblicza ich zawartości. I tu znów można otworzyć kolejne drzwi do nowego rozdziału poświęconego obliczeniom stężeń. Nasza maszyna oferuje dwie metody oznaczania stężeń: metodę podstawowych parametrów FPM oraz analizę w oparciu o krzywe kalibracyjne.
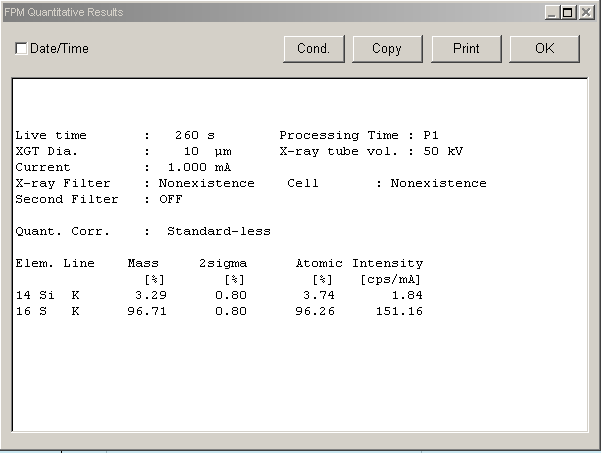
Rys. 15 Wynik ilościowy analizy obliczony metodą FPM.
Metoda FPM jest najprostsza i w wielu badaniach świetnie się sprawdza, nowoczesne oprogramowanie samo rozpoznaje położenia pików, ich intensywności i charakterystyczne wzorce z różnych linii emisji. Tak, jak powyżej, gdzie mamy wynik z badania czystej siarki. Urządzenie oprócz tego pierwiastka wykryło w nim 3,29% krzemu. W widmie mogą też być obecne pewne piki artefaktów, w tym charakterystyczne linie rozproszone Rayleigha i Comptona z generatora promieniowania rentgenowskiego, piki spowodowane dyfrakcją promieniowania rentgenowskiego oraz piki sumy/ucieczki. Ich znajomość jest konieczna, aby uniknąć błędnej interpretacji wyników. Dlatego uznaliśmy, że w tym artykule nie będziemy już głębiej wnikać w szczegóły a zrobimy to w kolejnych artykułach poświęconych konkretnym analizom, nie poruszymy tu już też drugiej metody obliczeń w oparciu o krzywe kalibracyjne, która zapewnia niesamowicie małe wykrywalności i oznaczania substancji od ppm do 100%.

Rys. 16 Przygotowanie próbki siarki do analizy.
Na przykładzie badania czystej siarki, którego wynik mogliśmy zobaczyć, przejdziemy do innego, acz bardzo ważnego w tych badaniach aspektu, mowa tu o preparatyce próbek. O ile z litymi metalami, ceramiką czy tworzywami sztucznymi nie ma problemu bo wystarczy niewielki kawałek takiego elementu położyć na stolik pomiarowy, o tyle w przypadku proszków lub materiałów niejednolitych musimy się bardziej postarać. Gdy chcieliśmy zbadać siarkę z naszego słoja z odczynnikiem nie mogliśmy od tak, wysypać jej odrobiny na stolik pomiarowy. Ciężko było by ustawić odpowiednią odległość głowicy pomiarowej no i wewnątrz maszyny jest spora ilość wentylatorów chłodzących poszczególne elementy, które po prostu zdmuchnęły by próbkę zanieczyszczając jednocześnie całą maszynę. W przypadku takich proszków można zastosować dwie metody preparatyki próbki: stapianie lub prasowanie.

Rys. 17 Przygotowanie próbki sproszkowanego złota.
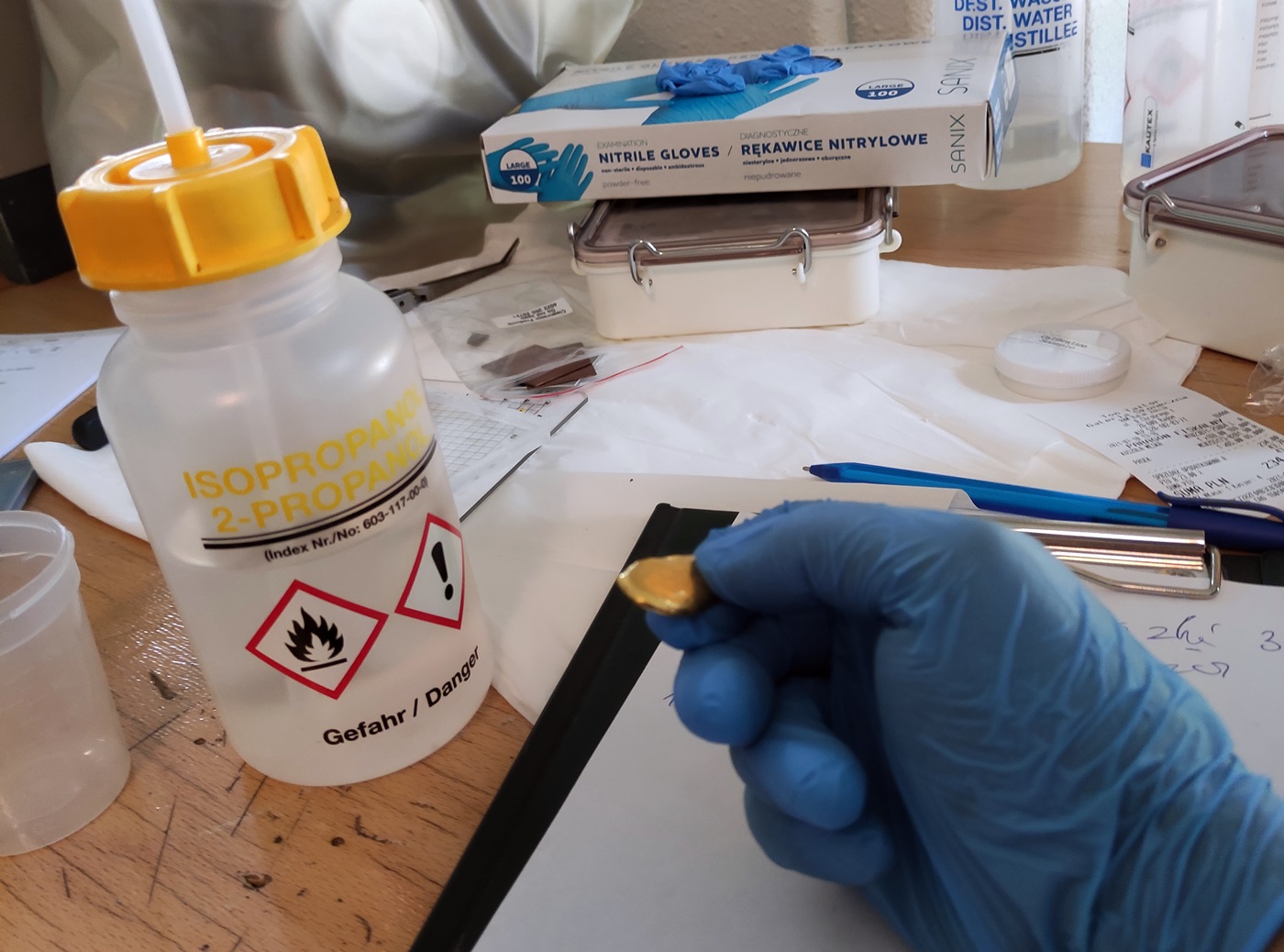
W przypadku siarki wystarczyło po prostu stopić próbkę co zapewniło jej zwartą i jednorodną strukturę, którą już w łatwy sposób można oświetlić wiązką. Jeśli stopienie jest nie możliwe, lub mogło by spowodować uszkodzenie materiału stosuje się prasowanie próbki do postaci tabletki. W tym przypadku klient dostarczył nam próbkę złota po chemicznym strąceniu z roztworu. Jego rozdrobnienie było niezwykle duże, najmniejszy podmuch powietrza powodował roznoszenie się pyłu.

Rys. 18 Proszek złota po chemicznym strąceniu z roztworu.
W takim przypadku możemy zastosować pastylkarkę oraz widoczną na poniższym zdjęciu hydrauliczną prasę zgniatającą o nacisku aż 100 ton. Nasza pastylkarka to urządzenie specjalnie wykonane do prasowania pastylek analitycznych pod wysokim ciśnieniem. Pomiędzy dwa walce wykonane z bardzo twardego spieku wsypuje się proszek i zgniata w stalowej tulei. Bardzo duże ciśnienie oddziałujące na materiał powoduje, że proszki zamieniają się w praktycznie jednolity, trwały materiał - idealny do badania naszą metodą. Jak widać na zdjęciu Nr 17 pastylka wykonana z proszku złota praktycznie niczym nie różni się od tłoczonej z metalu złotej monety.

Rys. 19 Hydrauliczna prasa o nacisku 100 ton.
Pracująca w naszym laboratorium prasa stosowana była do badań wytrzymałościowych betonu, jednak świetnie sprawdza się jako urządzenie do ściskania naszej pastylkarki. Jest to połączenie małego tłoczka, którym zwiększamy ciśnienie w tłoku zgniatającym o dużej średnicy, małym tłoczkiem trzeba się trochę namachać jednak dzięki temu uzyskiwane siły są bardzo duże. Jest to ważne gdy np. chcemy sprasować materiał nie mający chęci do połączenia się w jednolitą strukturę jak np.jedna z próbek osadu z komór do napylania metali naszego klienta, która w większości miała składać się ze srebra. W tym miejscu warto wspomnieć o metodzie uśredniania i przygotowania proszków do prasowania.

Rys. 20 Próbka zmielona w młynie planetarno - kulowym, przesiana i sprasowana.
W przypadku takich dziwnych i niejednolitych próbek do całej procedury preparatyki trzeba dodać jeszcze jedną lub dwie czynności. Punktowe pomiary mogły być bardzo nie miarodajne, gdyż takie osady zbierają się latami i na pewno nie są jednolite. Taką próbkę trzeba zmielić, przesiać, to co pozostanie na drobnym sicie jeszcze raz zmielić i znów przesiać i tak aż do ujednolicenia całej objętości próbki. Gdy mamy już jednorodny drobny proszek możemy z niego przygotować pastylkę analityczną jak powyżej.

Rys. 21 Młyn planetarno - kulowy do mielenia próbek.
Próbki można mielić na wiele sposobów, bardziej kruche ucieramy w agatowym moździerzu a dla bardziej opornych mamy młyn planetaro-kulowy. Młyny takie charakteryzują się bardzo szybkim i efektywnym mieleniem. Podczas pracy młyna naczynie obraca się wokół własnej osi i w kierunku przeciwnym, wokół osi przekładni obiegowej. Siły odśrodkowe, działające na ścianę naczynia powodują początkowo ruch kul zgodny z kierunkiem obrotu naczynia. Pojawią się różnice pomiędzy prędkością ścian naczynia i kul co powoduje powstawanie ogromnych sił tarcia, działających na próbkę. W miarę wzrastania szybkości obrotów, działające na kule siły Coriolisa, powodują ich odrywanie od ścian naczynia i uderzanie i zderzenia. Oderwane kule uderzają z ogromną siłą o próbkę znajdującą się po przeciwnej stronie naczynia. Powoduje to znaczną energię dynamiczną siły zderzenia.
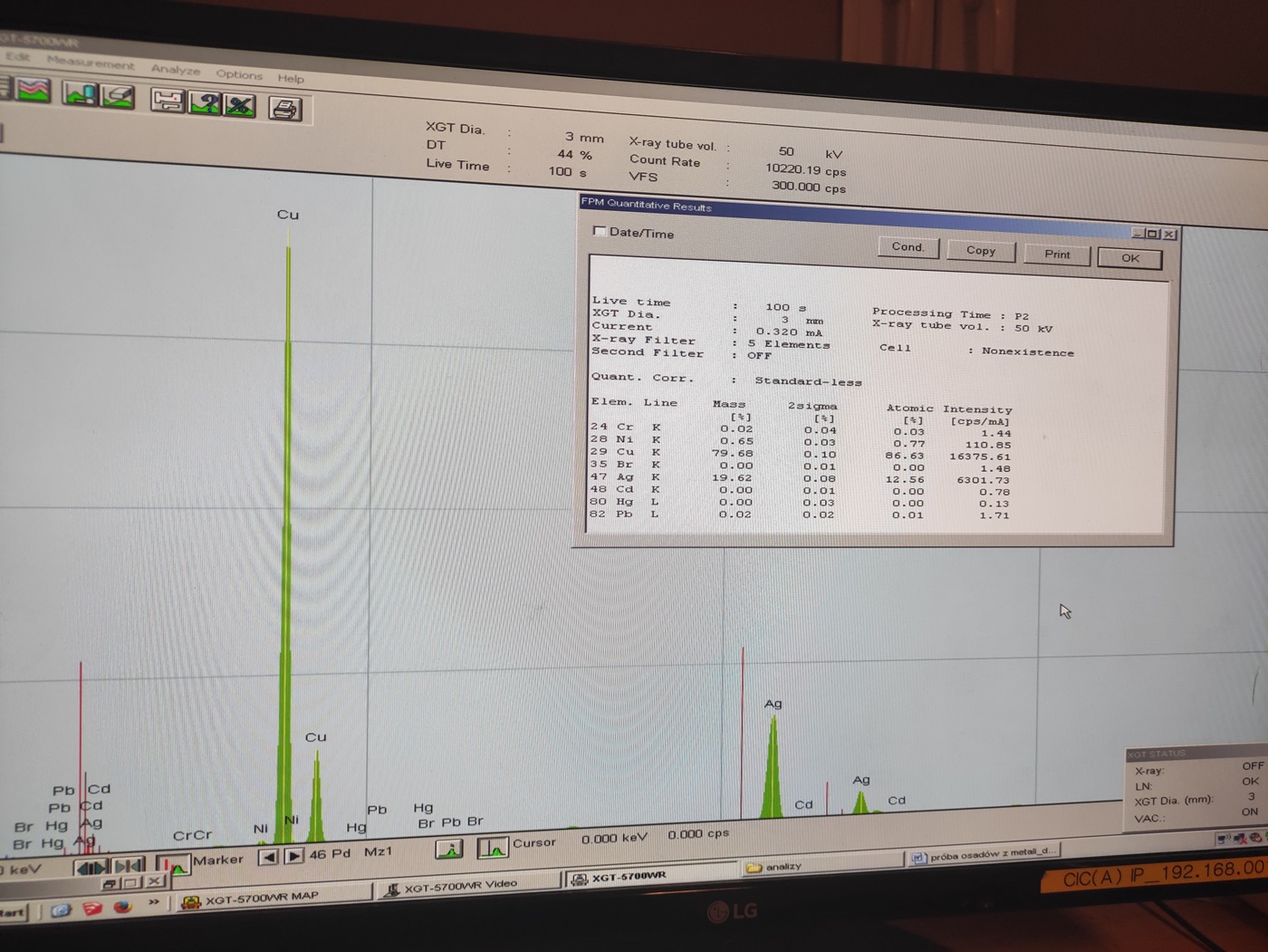
Rys. 22 Wynik analizy materiału, który w większości miał zawierać srebro.
W naszym młynie wykorzystujemy pojemniki i kule ze stali, ma to pewne konsekwencje dla przygotowywanej próbki. Otóż materiał ten, też ulega erozji w trakcie mielenia i nieznaczne ilości metalu przenikają do próbki powodując zafałszowanie późniejszego wyniku. Uwzględniamy to w późniejszym wyniku badania. W planach jest zakup naczynia i kul wykonanych z niezwykle twardego agatu lub korundu. Materiały te są tak odporne mechanicznie, że można śmiało mielić próbki bez obaw o zafałszowanie, jednak mają i swoją wadę a jest nią niezwykle wysoka cena.
Wracając jeszcze do próbki naszego klienta z materiałem z napylarki, nie był zadowolony z wyniku badania, które prezentujemy powyżej. Wg. nas i naszej maszyny srebro stanowiło jedynie 19,62% osadów a reszta to była głównie miedź. Klient uważał inaczej, jednak jak się później okazało mieliśmy całkowitą rację.

Rys. 23 Stapianie próbek metali w laboratoryjnym piecu muflowym.
Wspomnieliśmy wcześniej, że dostarczane nam próbki możemy również stopić, jeśli wiemy że mamy do czynienia z substancją, która nie ulegnie spaleniu, wyparowaniu lub wiemy na pewno, że wysoka temperatura w żaden sposób nie zaszkodzi ani próbce ani nam. Ta metoda jest szybsza, jednak wymaga znajomości jakościowej substancji a badanie ma na celu ustalenie składu procentowego składników. W naszej pracowni mamy do dyspozycji laboratoryjny piec muflowy z bardzo precyzyjnym sterowaniem cyfrowym, zapewniającym możliwość precyzyjnego zadania temperatury. Jest to ważne w pracach analitycznych, bo jeśli chcemy stopić dany metal, ceramikę lub inny materiał, którego skład z grubsza znamy, możemy tak dobrać temperaturę by np. nie przekroczyć w trakcie, temperatury parowania któregoś z nich.

Rys. 24 Stopiona próbka złota w trakcie stygnięcia.
I tu znów historia z naszych prac analitycznych na zlecenie. Klient dostarczył nam próbkę złota uzyskanego z recyklingu elektroniki. Było to również złoto w postaci proszku i tym razem postanowiliśmy je przetopić bo tak też umówiliśmy się z zamawiającym, gdy poinformował nas jak ostatnio próbował przetopić złoto palnikiem i większa jego część została wydmuchnięta z tygla.
Rys. 25 Próbka złota po przetopieniu w piecu muflowym, tzw. "gąska".
Być może w trakcie prac analitycznych oszczędziliśmy klientowi potencjalnych strat materiału. Nasza próbka metalu, którą prezentujemy powyżej, ważyła prawie 11g, dowiedzieliśmy się też że taki mały, nierównomierny odlew złota nosi nazwę "gąska". Po zabawie nią, przyszedł czas na naświetlenie jej promieniami rentgena i zarejestrowanie promieniowania fluorescencyjnego.
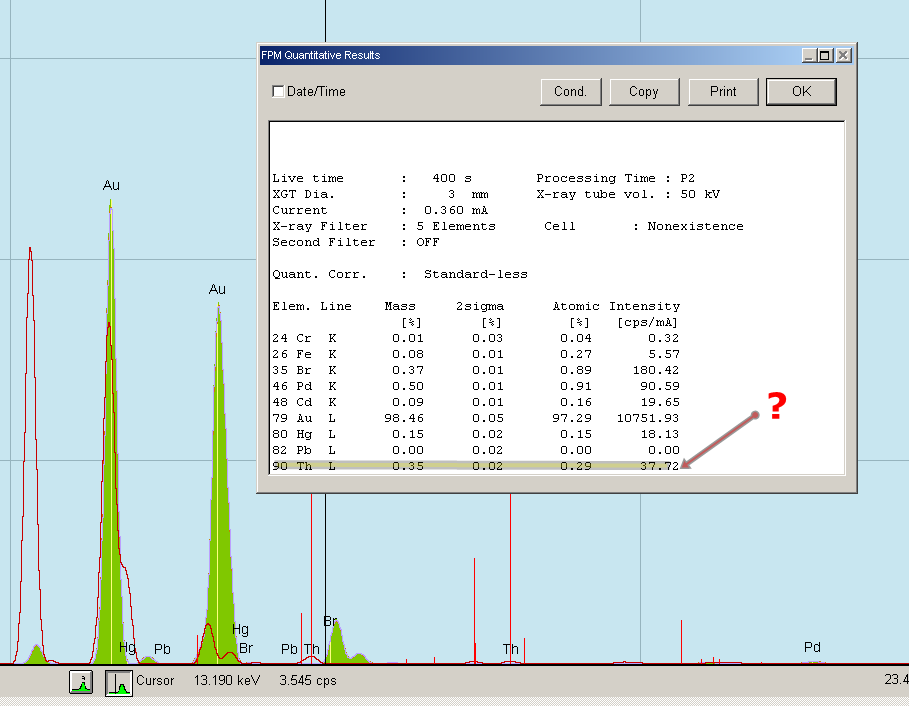
Rys. 26 Wynik badania stopionej próbki metalu.
Już na samym początku pomiaru wiedzieliśmy, że próbka w ogromnej większości składa się ze złota bo jego piki wyrosły "jak grzyby po deszczu", praktycznie natychmiast. Jest to dość ciężki pierwiastek i daje bardzo dobrą intensywność sygnału. Po zakończeniu pomiaru i obliczeniu wyniku metodą FPM mogliśmy stwierdzić, że próbka zawiera 98,46% czystego złota. Ten wynik zawiera także 5 pierwiastków z dyrektywy RoHS ( Cr, Br, Cd, Hg i Pb) dlatego, że w trakcie tego badania nie wiedzieliśmy jeszcze jak wyłączyć ten filtr i nie uwzględniać pierwiastków w wyniku, tzn. nie uwzględniać jeśli nie zostaną potwierdzone intensywnym sygnałem z ich kanałów energetycznych. W FPM z RoSH zawsze są w wyniku nawet jeśli ich sygnały są znikome.
W próbce wykryliśmy jeszcze niewielkie ilości żelaza, palladu no i toru. Ten ostatni pierwiastek nas i naszego klienta najbardziej zdziwił, bo jest to pierwiastek promieniotwórczy. Co prawda w próbce było go naprawdę niewiele, sprawdziliśmy ją nawet scyntylacyjnym detektorem promieniowania, jednak był w ilości 0,35%.
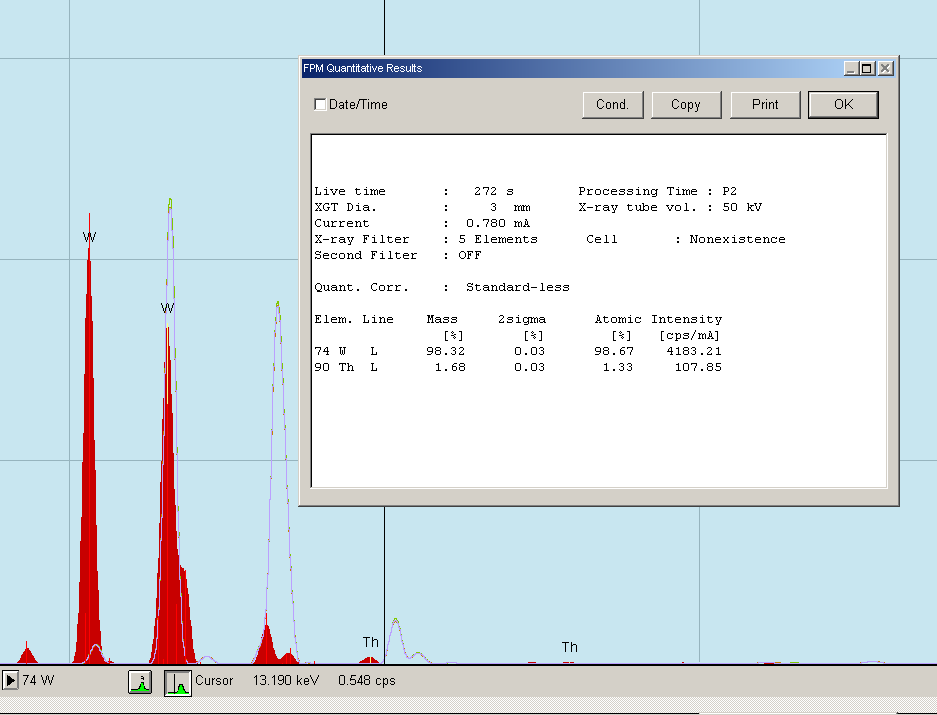
Rys. 27 Badanie referencyjne elektrody wolframowej z torem.
Zastanawialiśmy się czy nie jest to może jakiś błąd maszyny i tu przyszedł nam do głowy pomysł wrzucenia do analizy czegoś, co ten tor na pewno zawiera. Takim czymś była elektroda wolframowa do TIG-a (z czerwonym oznaczeniem, tzw. elektroda torowana), która zawiera ten pierwiastek w celu poprawy jonizacji łuku elektrycznego. Emitowane przez nią niewielkie promieniowanie jonizuje argon, który stanowi gaz osłonowy w tej metodzie spawania i sprawia że łuk elektryczny dużo lepiej się zajarza i jest stabilniejszy. Wykonaliśmy więc analizę tej elektrody i uzyskaliśmy wynik jak powyżej. Elektroda składała się z 98,32% wolframu i 1,68% toru.
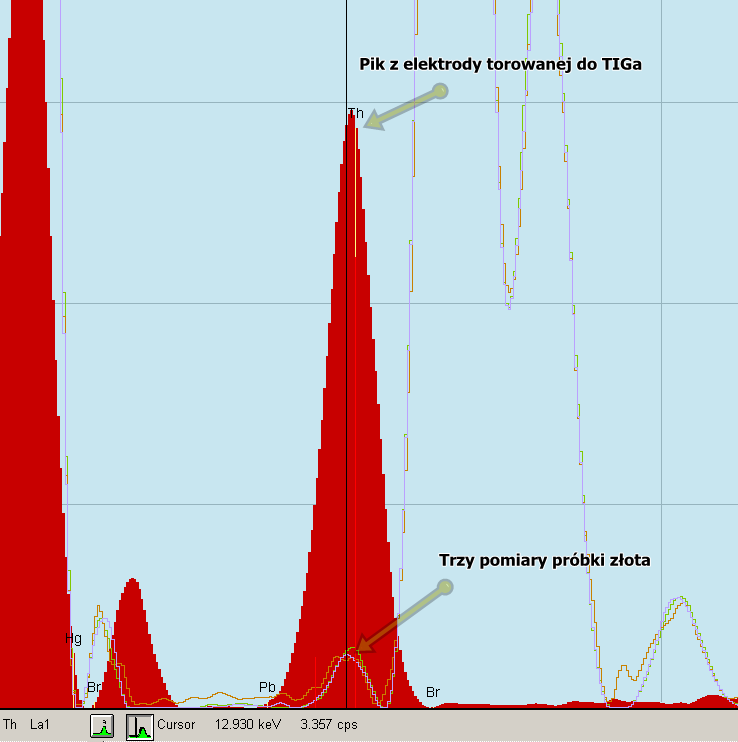
Rys. 28 Nałożenie badania referencyjnego na pomiar próbki.
Mając taki wynik, mogliśmy dzięki znakomitej funkcji programu, nałożyć widmo badania naszej próbki złota na widmo pomiaru referencyjnego elektrody. Na powyższym rysunku widzimy wynik tej operacji - duży czerwony pik to sygnał z badania elektrody a poniżej w tym samym miejscu mamy sygnały z trzech naświetlań naszej próbki złota. Sygnał z próbki złota bez wątpienia jest w paśmie energetycznym toru dlatego jasno mogliśmy stwierdzić, że nasza badana próbka na pewno go zawiera.
Analizując zastosowanie toru w w przemyśle znaleźliśmy informacje że jest on dodawany także do stopów styków z których wykonuje się styki przekaźników o bardzo dobrych parametrach łączeniowych. Często są to styki wykonane ze złota i srebra z dodatkiem właśnie toru. Z takich materiałów wykonuje się np. styki w przekaźnikach bezpieczeństwa. Jak się później okazało nasz klient odzyskiwał złoto m.in. z takich elementów i wyjaśniło się dlaczego jego próbka zawierała nieznaczne ilości tego pierwiastka. Była to merytoryczna wartość dodana w ramach usługi :).
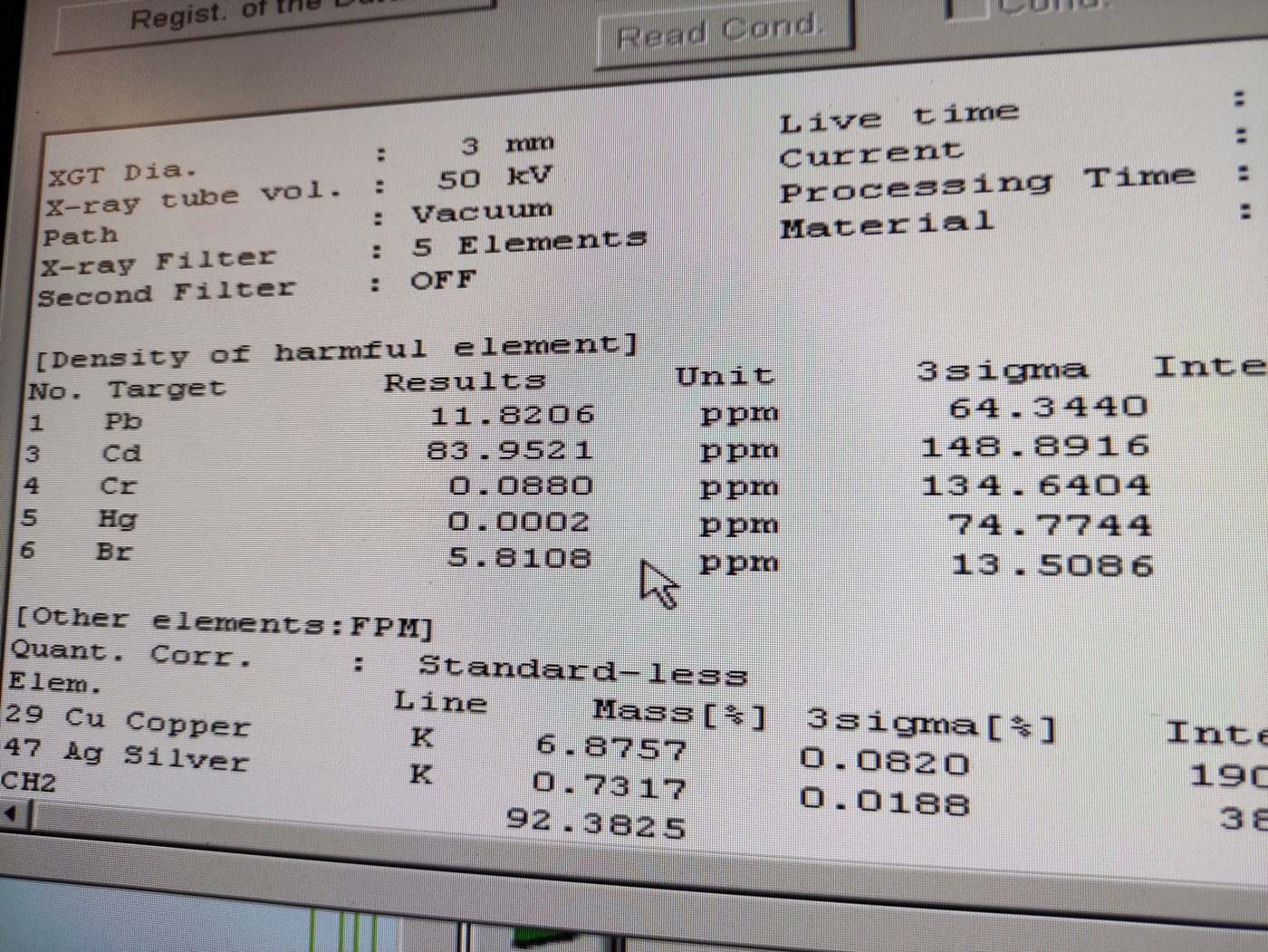
Rys. 29 Wynik badania w oparciu o krzywe kalibracyjne.
Na zakończenie tego artykułu powyżej prezentujemy jeszcze przykładowy wynik badania w oparciu o krzywe kalibracyjne. Jak wcześniej wspomnieliśmy tej metodzie poświęcimy oddzielne opracowanie. Jednak wykonaliśmy taką próbę w oparciu o krzywe już wcześniej przygotowane w systemie. Są to krzywe kalibracyjne wykonane dla badania RoHS dla pięciu niebezpiecznych pierwiastków. Zawartości podane są w jednostkach ppm ( parts per million). Oczywiście można też prowadzić analizę każdej próbki także dla jednego pierwiastka, dla którego została wykonana krzywa w konkretnej matrycy, np. w plastiku, biomasie czy ceramice.

Rys. 30 Akcesoria kalibracyjne maszyny XRF.
Do wykonania takiej krzywej kalibracyjnej konieczne są odpowiednie wzorce. Sporą ilość takich wzorców kupiliśmy wraz z maszyną, jednak ich ogromna większość związana jest z badaniem RoHS w różnych materiałach. Dla naszych analiz będziemy musieli wykonać własne lub zakupić fabryczne. Te działania będą też uzależnione od zapotrzebowania na konkretne badania. W ostatnim czasie zwrócił się do nas firma z zapytaniem o szybkie badanie większej ilości próbek mikrosfer ceramicznych pochodzących z popiołów piecowych na zawartość chloru. Takie mikrosfery wykorzystuje się do produkcji materiałów budowlanych, a chlor w materiałach budowlanych jest bardzo niepożądany i jego zawartość determinuje jakość. W budownictwie stosuje się dużo wapnia a ten pierwiastek tworzy z chlorem bardzo higroskopijny chlorek wapnia. Raczej nikt by nie chciał by po jakimś czasie cegły zaczęły się rozpływać. Będziemy musieli przygotować preparatykę próbek, przygotować krzywą kalibracyjną w takim materiale i wreszcie wycenić badanie zakładając, że współpraca będzie stała. Wykonanie takiej pojedynczej analizy, ze względu na te wszystkie czynności tą metodą było by po prostu nie opłacalne.
Wyciągając wnioski, możemy jasno stwierdzić, że jest to naprawdę wygodna, szybka, nieinwazyjna i pewna metoda analizy materiałowej, jednak dla zapewnienia opłacalności badania, wymagane jest zapewnienie ciągłości powtarzalnych badań. Dlatego, tego typu maszyny, są tak popularne w laboratoriach kontroli jakości zakładów produkcyjnych gdy potrzeba badać grupę stałych parametrów w tych samych materiałach.
W naszym laboratorium będziemy mieli trochę więcej problemów, by zapewnić konkretne badania w przyzwoitej cenie, jednak biorąc pod uwagę też badania własne, cele edukacyjne i inne pośrednie badania jak np. oznaczanie siarki w paliwach do badania ich kaloryczności, może się udać.










